Postmortem: RIG: Recalibration and Interrelation of Genomic Sequence Data with the GATK
As mentioned in the previous post, the Llama and I recently got a workflow for using the GATK with a non-model organism published in G3: "RIG: Recalibration and Interrelation of Genomic Sequence Data with the GATK", and it's postmortem time (the code base can be found on GitHub here).
This manuscript was mostly a product of fortunate timing. Around early 2013, The Llama and I had been accepted as students in the lab we're currently in, and one of the things we were given to work on was converting restriction enzyme based reduced representation sequence data (a.k.a. Digital Genotyping or RAD-seq) to genotypes for the purpose of genetic map construction and QTL mapping. After iterating through a few ideas, we found that the software Stacks would be a good fit for our use case. We used Stacks for a little while, and its reference genome based pipeline worked fairly well for us.
Somewhere around mid to late 2013 we needed to do some variant calling from whole genome sequence data. Previously, CLC Genomics Workbench had been used by the lab for this application. However, the Llama and I go out of our way to avoid non-open source software, and we looked for alternatives. We caught wind of the GATK and its many blessings, and started to learn the ropes for whole genome sequence variant calling. For the time capsule, here's a forum post where I asked the GATK forum for advice in generating a variant set for BQSR/VQSR, and here's a forum post where I asked the Stacks forum for advice on generating a VCF file for use in recalibration with the GATK; that Stacks post was the nascent form of what ultimately evolved into the RIG workflow.
Since we had a reference genome, we transitioned away from Stacks and exclusively used GATK. This allowed us to use one tool that would produce standard file formats for calling variants from both our RR and WGS data; this made it much easier to identify the same variants from both types of data in downstream analyses. So, we started using the UnifiedGenotyper for large sample numbers of RR biparental family and association population analyses, and the HaplotypeCaller for the small number of WGS samples.
Then came the GATK 3.0 release which brought the power of the HaplotypeCaller to bear on as many samples as desired thanks to the new database of likelihoods model. By this time we had something like 6 full sequencing runs (48 lanes) of RR sequence data and were running into scaling problems trying to run the analyses on a local workstation. To get around this, we decided to invest some time porting our current analysis pipeline to use the GATK's Queue framework on a university high performance computing cluster.
For initial testing, we set up Sun Grid Engine on a single workstation (setting it as both a submission and execution host; is that weird?) to prototype pipelining in Scala with Queue and got our whole pipeline up and running beautifully on the single machine: Queue was happily Scatter-Gathering and HaplotypeCaller was churning through 100's of samples in reasonable timescales (although not fast enough that it would be sustainable to not move to a compute cluster). It was awesome.
Unfortunately it wasn't so awesome on the compute cluster. We ran into a problem where Queue didn't interact with the resource manager (Univa Grid Engine) nicely, and the job submissions slowed to a crawl over time:

...

After much consternation, some forum posts, and lots of things that didn't work, we eventually threw in the towel. While I suspect that figuring it out would have been instructive from a computing standpoint, there were data that needed to be analyzed. We figured it would be safer to do a rewrite in Bash that we knew would work rather than try to debug the interaction between our Queue pipeline and the cluster architecture, so we threw away the Scala code and rewrote the pipeline in Bash. Unfortunately, we had to recreate some of what made Queue so valuable, including logging and dependency tracking, but at least it was working on the cluster.
Around that time, we were designing the first iterations of what would become the RIG workflow. Back then it was internally called the LERRG workflow (Leveraging Existing Resources for Recalibration with the GATK). Say it out loud.
LERRG.
It sounds like something you might cough up in the morning, and it looked similarly awful:
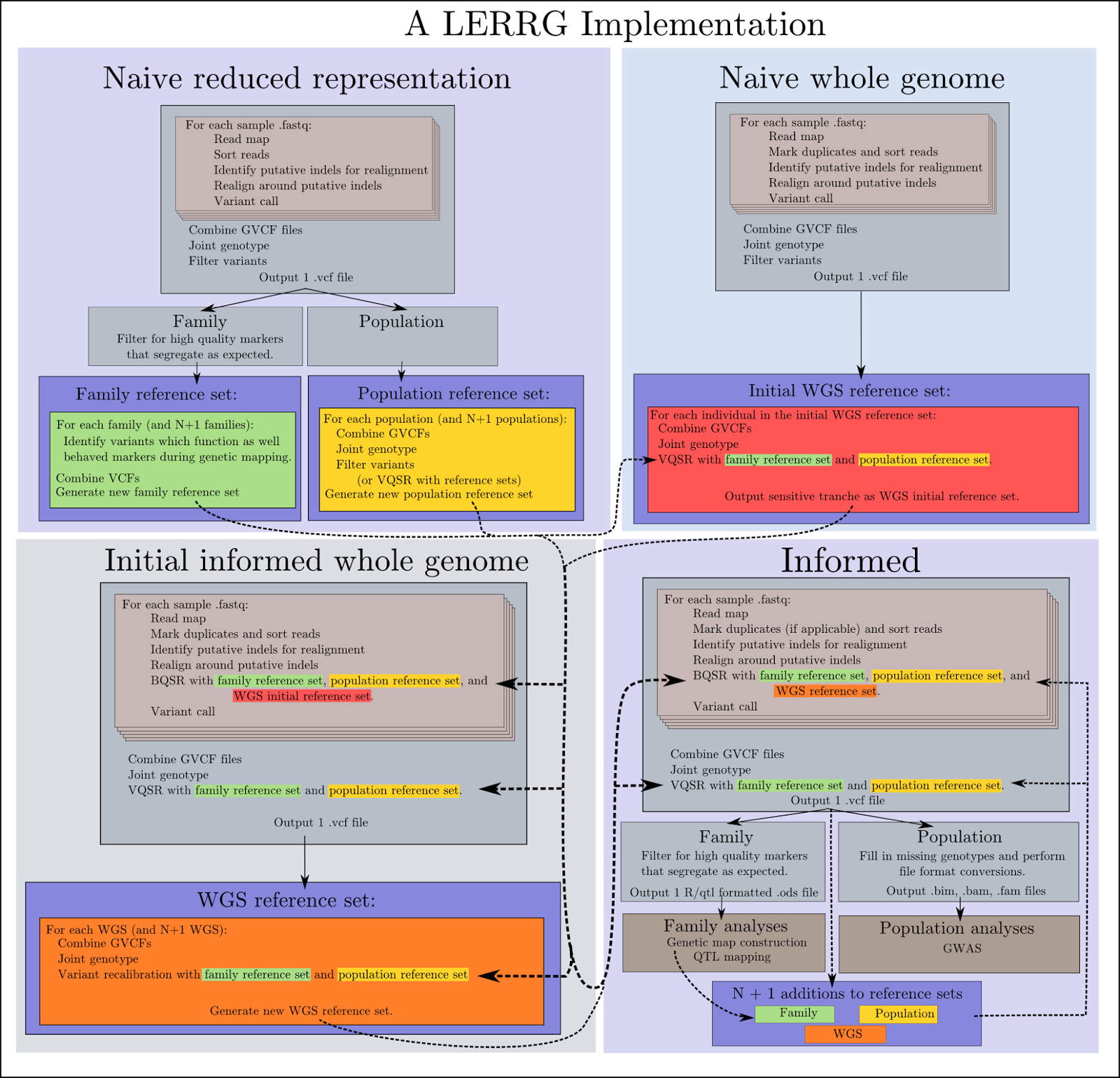
The basic premise was to use variants called from RR data to inform VQSR of WGS data, and then to use the recalibrated WGS variants for BQSR of the WGS reads, followed by another round of VQSR after variant calling from the BQSRed WGS reads.
Once we had the idea and code in place, it was just a matter of execution. As expected, the workflow works pretty well; the GATK's methods are solid so it would have been surprising if it didn't work. It also worked just fine to recalibrate the RR reads and variants.
We started to kick around the idea of submitting the workflow as a manuscript since it looked like there was a market for it given that the GATK forum often has questions on how to use the GATK in non-model organisms (e.g. here and here), and the workflow was too complex to try to keep describing it in the methods sections of other papers from the lab. So, we wrote up the manuscript; during internal revisions it got renamed to RIG: Recalibration and Interrelation of genomic sequence data with the GATK. It also took considerable effort on the Llama's part to convince me that the diagram I had constructed for LERRG was, in fact, terrible; I doubt the manuscript would have ever been accepted had she not intervened and redesigned the thing from the ground up as we were working on the manuscript.
After a rapid reject from Molecular Plant, we targeted G3 since the review process there has been fair and reasonably fast in the past, and G3's readership seems to have a good number of communities that were in similar situations to ours: genomics labs working on organisms with a reference genome and a sizeable chunk of sequence data. The reviews we got back suggested testing the workflow in a better characterized system, and so we benchmarked it in Arabidopsis with good results.
What worked:
Timing: This was mostly a case of being in the right place at the right time. The GATK's 3.0 release occurred just as we were scaling up and in position to take full advantage of it, and our internal analysis pipeline hadn't been locked in so we were free to test out different options. The GATK had already been widely adopted by the human genomics community, and interest was growing in other communities in applying its benefits.
Prototyping, Refactoring, and Iteration: This was a relatively small scale project without many cooks in the kitchen, and many iterations of prototyping, testing, and redesigning or refactoring worked well. I doubt it would work well on a larger project, but the agility let us learn and improve things as we went.
What didn't work:
Development environment didn't represent deployment environment: We wasted a good chunk of time trying to debug why the Queue pipeline we wrote worked locally with Sun Grid Engine but not on the compute cluster with Univa Grid Engine. My lack of understanding of the internals of the resource manager and Queue prevented me from ever getting to the bottom of it, but we would have known about it much sooner if we had started developing and testing on the compute cluster where we would actually be running the analyses. Fortunately, we still learned something about resource managers by setting up SGE locally and learned a bit about Scala from the prototype.
Slow test cases: I still haven't generated any rapid test cases for testing and development of the workflow. Unfortunately, many of the problems arise mostly as a consequence of scale (e.g. keeping too many intermediate files consume hard drive space allocation before the pipeline finishes; file handle limit too small on the compute cluster; non-deterministic thread concurrency crashes with parallelized HaplotypeCaller). I haven't found a way to test for these without running an analysis of a full dataset, and that can require hours before the crash occurs; simple debugging takes days sometimes.
This manuscript was mostly a product of fortunate timing. Around early 2013, The Llama and I had been accepted as students in the lab we're currently in, and one of the things we were given to work on was converting restriction enzyme based reduced representation sequence data (a.k.a. Digital Genotyping or RAD-seq) to genotypes for the purpose of genetic map construction and QTL mapping. After iterating through a few ideas, we found that the software Stacks would be a good fit for our use case. We used Stacks for a little while, and its reference genome based pipeline worked fairly well for us.
Somewhere around mid to late 2013 we needed to do some variant calling from whole genome sequence data. Previously, CLC Genomics Workbench had been used by the lab for this application. However, the Llama and I go out of our way to avoid non-open source software, and we looked for alternatives. We caught wind of the GATK and its many blessings, and started to learn the ropes for whole genome sequence variant calling. For the time capsule, here's a forum post where I asked the GATK forum for advice in generating a variant set for BQSR/VQSR, and here's a forum post where I asked the Stacks forum for advice on generating a VCF file for use in recalibration with the GATK; that Stacks post was the nascent form of what ultimately evolved into the RIG workflow.
Since we had a reference genome, we transitioned away from Stacks and exclusively used GATK. This allowed us to use one tool that would produce standard file formats for calling variants from both our RR and WGS data; this made it much easier to identify the same variants from both types of data in downstream analyses. So, we started using the UnifiedGenotyper for large sample numbers of RR biparental family and association population analyses, and the HaplotypeCaller for the small number of WGS samples.
Then came the GATK 3.0 release which brought the power of the HaplotypeCaller to bear on as many samples as desired thanks to the new database of likelihoods model. By this time we had something like 6 full sequencing runs (48 lanes) of RR sequence data and were running into scaling problems trying to run the analyses on a local workstation. To get around this, we decided to invest some time porting our current analysis pipeline to use the GATK's Queue framework on a university high performance computing cluster.
For initial testing, we set up Sun Grid Engine on a single workstation (setting it as both a submission and execution host; is that weird?) to prototype pipelining in Scala with Queue and got our whole pipeline up and running beautifully on the single machine: Queue was happily Scatter-Gathering and HaplotypeCaller was churning through 100's of samples in reasonable timescales (although not fast enough that it would be sustainable to not move to a compute cluster). It was awesome.
Unfortunately it wasn't so awesome on the compute cluster. We ran into a problem where Queue didn't interact with the resource manager (Univa Grid Engine) nicely, and the job submissions slowed to a crawl over time:
...
After much consternation, some forum posts, and lots of things that didn't work, we eventually threw in the towel. While I suspect that figuring it out would have been instructive from a computing standpoint, there were data that needed to be analyzed. We figured it would be safer to do a rewrite in Bash that we knew would work rather than try to debug the interaction between our Queue pipeline and the cluster architecture, so we threw away the Scala code and rewrote the pipeline in Bash. Unfortunately, we had to recreate some of what made Queue so valuable, including logging and dependency tracking, but at least it was working on the cluster.
Around that time, we were designing the first iterations of what would become the RIG workflow. Back then it was internally called the LERRG workflow (Leveraging Existing Resources for Recalibration with the GATK). Say it out loud.
LERRG.
It sounds like something you might cough up in the morning, and it looked similarly awful:
The basic premise was to use variants called from RR data to inform VQSR of WGS data, and then to use the recalibrated WGS variants for BQSR of the WGS reads, followed by another round of VQSR after variant calling from the BQSRed WGS reads.
Once we had the idea and code in place, it was just a matter of execution. As expected, the workflow works pretty well; the GATK's methods are solid so it would have been surprising if it didn't work. It also worked just fine to recalibrate the RR reads and variants.
We started to kick around the idea of submitting the workflow as a manuscript since it looked like there was a market for it given that the GATK forum often has questions on how to use the GATK in non-model organisms (e.g. here and here), and the workflow was too complex to try to keep describing it in the methods sections of other papers from the lab. So, we wrote up the manuscript; during internal revisions it got renamed to RIG: Recalibration and Interrelation of genomic sequence data with the GATK. It also took considerable effort on the Llama's part to convince me that the diagram I had constructed for LERRG was, in fact, terrible; I doubt the manuscript would have ever been accepted had she not intervened and redesigned the thing from the ground up as we were working on the manuscript.
After a rapid reject from Molecular Plant, we targeted G3 since the review process there has been fair and reasonably fast in the past, and G3's readership seems to have a good number of communities that were in similar situations to ours: genomics labs working on organisms with a reference genome and a sizeable chunk of sequence data. The reviews we got back suggested testing the workflow in a better characterized system, and so we benchmarked it in Arabidopsis with good results.
What worked:
Timing: This was mostly a case of being in the right place at the right time. The GATK's 3.0 release occurred just as we were scaling up and in position to take full advantage of it, and our internal analysis pipeline hadn't been locked in so we were free to test out different options. The GATK had already been widely adopted by the human genomics community, and interest was growing in other communities in applying its benefits.
Prototyping, Refactoring, and Iteration: This was a relatively small scale project without many cooks in the kitchen, and many iterations of prototyping, testing, and redesigning or refactoring worked well. I doubt it would work well on a larger project, but the agility let us learn and improve things as we went.
What didn't work:
Development environment didn't represent deployment environment: We wasted a good chunk of time trying to debug why the Queue pipeline we wrote worked locally with Sun Grid Engine but not on the compute cluster with Univa Grid Engine. My lack of understanding of the internals of the resource manager and Queue prevented me from ever getting to the bottom of it, but we would have known about it much sooner if we had started developing and testing on the compute cluster where we would actually be running the analyses. Fortunately, we still learned something about resource managers by setting up SGE locally and learned a bit about Scala from the prototype.
Slow test cases: I still haven't generated any rapid test cases for testing and development of the workflow. Unfortunately, many of the problems arise mostly as a consequence of scale (e.g. keeping too many intermediate files consume hard drive space allocation before the pipeline finishes; file handle limit too small on the compute cluster; non-deterministic thread concurrency crashes with parallelized HaplotypeCaller). I haven't found a way to test for these without running an analysis of a full dataset, and that can require hours before the crash occurs; simple debugging takes days sometimes.


Comments
Post a Comment