Skip to main content
Search
Search This Blog
Codex Technicanum
We have no idea what that means.
Home
The Scientists
Credentials
More…
Posts
Showing posts from July, 2013
Show all
Posted by
Llama
July 30, 2013
Rename error in Linux VirtualLeaf tutorial: "Bareword "tutorial0" not allowed while "strict subs" in use at (eval 1) line 1."
Posted by
Frogee
July 30, 2013
Nature News article: Quantum boost for artificial intelligence
Posted by
Frogee
July 29, 2013
Beginnings with Git
Posted by
Frogee
July 29, 2013
Advice from a systems software engineer
Posted by
Llama
July 26, 2013
compile errors in Linux VirtualLeaf installation
Posted by
Frogee
July 25, 2013
Configuring an IGV Genome Server
Posted by
Frogee
July 23, 2013
On the number of markers and the number of individuals
Posted by
Frogee
July 22, 2013
Determine size of folders and files in directory
Posted by
Frogee
July 21, 2013
Extracting sequence information from Stacks samples
Posted by
Frogee
July 18, 2013
Search and replace in a file without opening it
Posted by
Frogee
July 17, 2013
Apache error log location in Linux Ubuntu 12.10
Posted by
Frogee
July 15, 2013
Compression of files in parallel using GNU parallel
Posted by
Frogee
July 13, 2013
In silico restriction digest and retrieval of restriction site associated DNA
Posted by
Frogee
July 12, 2013
Determine the type of an object in Python
Posted by
Frogee
July 12, 2013
Call external program from Python and capture its output
Posted by
Frogee
July 11, 2013
Installation of Biopieces on Ubuntu Linux 12.10 - Ruby installation
Posted by
Frogee
July 11, 2013
In silico restriction enzyme digest of eukaryotic genome
Posted by
Frogee
July 10, 2013
Count number of reads in a SAM file above or below a mapping quality score with awk
Posted by
Frogee
July 09, 2013
Find CPU information in Ubuntu (12.10)
Posted by
Frogee
July 09, 2013
Interesting response to Nature Article "Biology must develop its own big data systems"
Posted by
Frogee
July 09, 2013
Read in file by command line argument and split on tab delimiter in Python
Posted by
Llama
July 08, 2013
$\LaTeX$ in LibreOffice Impress
Posted by
Frogee
July 08, 2013
Newbie awk usage example
Posted by
Frogee
July 08, 2013
Vim regex
Posted by
Frogee
July 08, 2013
How to show line numbers in vim
Posted by
Frogee
July 08, 2013
Write ordered list in LaTeX using enumerate
Posted by
Frogee
July 08, 2013
How to show LaTex in Blogger
Posted by
Frogee
July 07, 2013
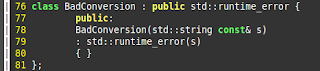
How to show code snippets in Blogger
Posted by
Llama
July 07, 2013
smart and graduate studies are mutually exclusive
Posted by
Frogee
July 07, 2013
Collaborative meeting platform
Posted by
Frogee
July 07, 2013
Count number of files in directory using bash
Newer Posts
Home